研究背景
材料科学在现代科技中起到基础性的关键作用。传统的材料研发模式主要通过优化实验参数以获得最佳材料,面临长周期、高成本、低能效等挑战,无法满足21世纪工业发展对新型材料的需求。然而,随着材料理论计算模拟技术的飞速发展,材料研发模式正逐步从实验驱动向计算驱动转变,并且在人工智能(AI)的助力下,正迈向数据驱动的新阶段。以理论计算为基石,通过高通量计算技术迅速获取海量数据,随后利用机器学习与数据挖掘技术,基于这些大数据集构建预测模型,探寻潜在的新型材料。这一创新方法借助人工智能,极大地加速了新型材料的筛选与设计进程,同时显著降低了研发成本。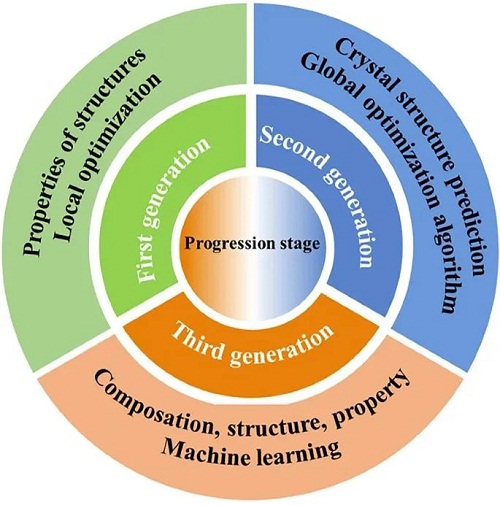
Artificial Intelligence‑Powered Materials Science
Xiaopeng Bai, Xingcai Zhang*
Nano-Micro Letters (2025)17: 135
https://doi.org/10.1007/s40820-024-01634-8
本文亮点
1. 全面解析人工智能与机器学习技术在材料研发中的创新应用。
2. 深入探讨人工智能驱动材料科学的关键难题与突破方向。
3. 通过前沿案例,展现人工智能如何加速材料开发与发现,开启材料科学新纪元。
内容简介
材料的进步是推动人类文明发展的核心动力。AI驱动的材料科学预示着新时代的到来,在能源、环境和生物医学等领域,贡献环保、高效、益民的先进技术方案。斯坦福大学张兴才等归纳了数据驱动的新型AI材料开发流程,围绕碳基功能材料、无机材料及复合材料三大类,深入剖析了AI驱动的材料科学进展与前沿应用,展望了AI技术在材料研发领域的广阔应用前景,同时提出该领域面临的机遇与挑战。AI将成为材料创新的加速器,而材料创新的成果又将反哺AI预测能力的提升,促进AI与材料科学的共生发展。两者协同并进,有望开启一个由先进AI材料驱动的未来。
图文导读
I 人工智能与材料研发
人工智能通过模拟人类智能并利用数据驱动技术,超越了自然智能在速度、效率和生产力方面的能力,从而加速了新材料等领域的研发与生产。机器学习是人工智能的子领域,通过数据驱动技术训练模型预测并解决任务,通常包括描述符生成、模型构建与验证、材料预测和实验验证等四个步骤。一方面,机器学习有效利用材料科学的大数据,建立准确预测模型,充分挖掘数据潜力。另一方面,计算材料学中的传统模拟方法耗时耗力且存在局限,而机器学习则能高效识别复杂的材料特性关系,显著减少计算时间并扩展系统尺度。与传统的试错法相比,机器学习技术具有成本低、效率高、周期短和可扩展性等优势,且能仅基于数据以高度非线性的形式训练模型,不依赖物理原理,因此它已成为材料科学中预测特性、筛选和优化设计的重要工具。
1.1 材料模拟的发展阶段
通过计算模拟预测材料特性的方法分为三个发展阶段。第一代方法通过近似求解薛定谔方程和局部优化技术计算材料的物理特性;第二代方法利用全局优化算法,基于成分预测材料结构;第三代方法则采用机器学习,依赖充足实验数据预测材料的成分、结构和特性。第三代方法要求构建以数据为核心,整合高通量实验、高通量计算和材料数据的综合性创新平台。因此,新型AI材料的开发流程包含三个主要阶段:通过现有材料的表征和计算平台获取数据、利用AI模型进行数据分析,以及基于识别出的数据特征生成新材料。
1.2 机器学习数据库
机器学习发展需大量实验数据,目前,现有的数据库如表1所示。
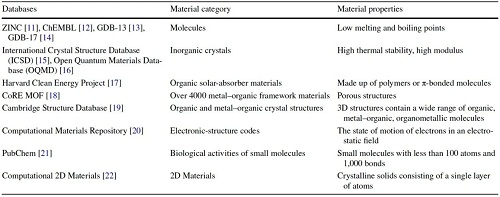
表1. 材料领域常用数据库
ZINC8专注于提供化合物信息,助力药物发现;ChEMBL汇集小分子生物活性数据,促进SAR研究和靶标验证;GDB-13和GDB-17系统枚举小有机分子,拓展化学空间;ICSD收录实验确定的晶体结构,为晶体学研究提供宝贵资源;OQMD聚焦材料信息学和量子力学计算,推动高通量材料筛选;CEP致力于发现和设计太阳能有机材料;CoRE MOF汇编实验表征的MOFs,促进新型MOFs设计;CSD提供小分子和金属-有机晶体结构信息,助力结构研究;CMR存储和分享计算材料科学数据,推动新材料发现;PubChem整合多源化学信息,支持药物发现和化学生物学研究;计算二维材料数据库关注二维材料性质,助力新型材料设计和优化。这些机器学习数据库对药物发现、晶体学和计算建模等领域至关重要,它们提供丰富的化学和材料数据,助力研究人员探索化学空间、分析结构-活性关系、设计新材料,加速创新解决方案的发现。
1.3 机器学习算法模型
在收集到所需数据后,研究人员需将数据转换为机器可理解的格式,并利用TensorFlow、Keras和Scikit-Learn等开源AI框架开发机器学习模型。这些框架提供灵活性、可定制性和社区交流合作。机器学习依赖于多种算法,分为监督、无监督和半监督学习,其中机器学习(ML)算法如SVMs和决策树适用于结构化数据,而深度学习(DL)模型如ANNs更适合复杂、高维数据。部署DL模型需更多数据和计算资源,并面临数据标准化、模型可解释性和计算效率等挑战。
监督学习包括决策树、朴素贝叶斯、支持向量机、线性回归、K最近邻、核岭回归和人工神经网络等常用算法,用于分类和回归任务,能够处理线性和非线性关系。无监督学习如聚类和特征降维,缺乏正确答案指导,自主发现和揭示数据结构,自编码器是其一种神经网络架构。半监督学习结合标记和未标记数据,可用于应对获取大量标记数据的挑战。这些算法在材料信息挖掘中具有潜力,但需进一步开发开源算法。然而,机器学习模型的有效性取决于数据质量,因此需解决数据表示、相关性和数据不足等问题,促进机器学习方法在材料科学中的更广泛应用。
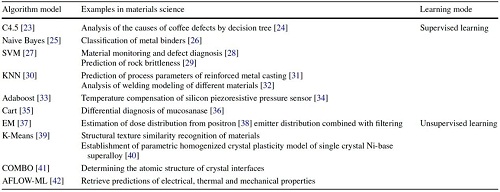
表2. 材料科学中常用的机器学习算法
II AI驱动的材料开发
近年来,人工智能在材料科学领域的应用激增,材料大致可分为三大类(图1):碳基功能材料(有机材料,如碳纳米管和有机发光二极管)、无机材料(如贵金属纳米粒子和二维材料)和复合材料(如金属有机骨架材料)。这些材料在多个领域展现出广泛的应用性,包括电子、医学、图像处理、生物医学、智能机器人和电催化等。人工智能在材料科学中的作用主要分为材料发掘、性能预测、优化与设计,以及过程模拟与制造。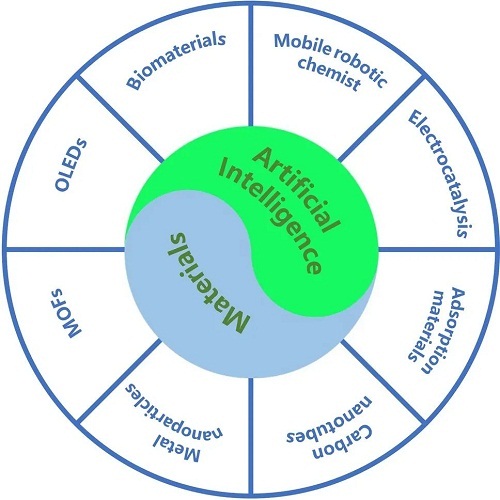
图1. 人工智能驱动的材料分类
2.1 材料发掘
碳纳米管具有高强度、高刚度、优异导电性和导热性,应用广泛但合成面临挑战。人工智能在碳纳米管合成中发挥变革性作用,如Nikolaev等人的研究中设计的基于人工智能的自主研究系统(ARES),通过机器学习自主实验并优化合成,其迭代闭环系统使合成过程比传统方法更快。ARES系统通过自动化实验设计,以迭代方式自主执行超过600次实验,大大加快了碳纳米管(CNT)的合成进程。它持续学习并不断完善预测,随着实验次数增加,预测生长率与实际生长率差异逐渐减小。ARES能够探索复杂的参数空间,利用机器学习在多维参数中导航,快速识别最优合成条件。系统还集成了原位检测与表征功能,实现实时监测和即时调整,确保更好合成结果。随着数据库扩大和实验次数增加,ARES准确性提高,减少了达到期望结果的试验次数。图2a-2e展示了这一过程:实验生长率与预测生长率随实验次数增加而收敛(图2a、2b);模拟实验参数生长的CNT与观察生长率成正比(图2c);有限且稀疏数据集导致参数选择范围广泛,成功率低(图2d);数据集增大后,预测与实验差距缩小,成功率显著提高(图2e)。该工作强调了机器学习在材料研究中的革命性潜力,展示了其在推动新发现方面的作用,并预示了自主材料研究的未来发展趋势。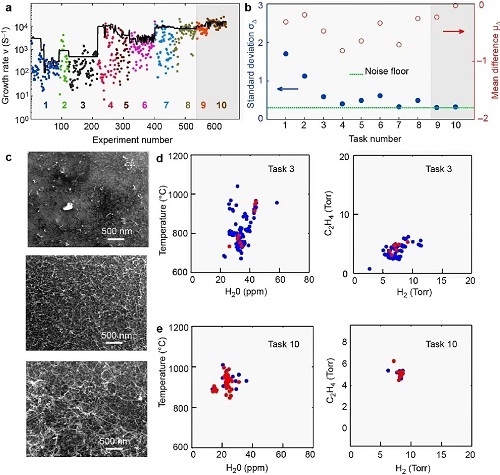
图2. ARES用于单壁碳纳米管合成的研究。(a) 实验生长速率与ARES预测生长速率的比较;(b) 通过量化实验观测到的生长速率与预测生长速率之间的差异来评估收敛性;(c) 基于模拟实验参数合成的碳纳米管;(d) 使用有限且稀疏的数据集进行训练的模型,导致参数选择范围广泛;(e) 模型的数据集比图2d所示的数据集大三倍,从而使得预测结果与实验结果之间的差异更小。
2.2 材料性能预测
双金属或多金属材料因其合金化金属的协同效应广泛应用于催化化学和电化学反应中。然而,发现合适的合金材料耗时且昂贵。人工神经网络等机器学习技术在快速准确预测吸附能、催化活性和选择性等性能方面显示出巨大潜力。Cu对CO₂具有显著的电还原活性,设计(100)晶面封端的双金属或多金属材料对提高CO₂还原为C₂物种的效率有重要意义。Ma等人提出机器学习增强的化学吸附模型,能在广泛化学空间内快速准确预测金属合金的表面反应性。该模型建立了材料特征与CO吸附能之间的非线性映射,高通量计算关键描述符,预测和理解催化活性、最佳组成和活性位点。图3a-3e展示了CO₂电还原过程中C1和C₂物种的自由能路径、理论极限电位、Cu₃B-A@Cu单层表面CO吸附能的筛选、神经网络模型与密度泛函理论(DFT)计算的比较,以及主要特征与主体金属M之间的关系。此外,Tran等人开发了基于代理优化和主动机器学习的工作流程,筛选了1499种金属间化合物,证实了多种具有优异CO₂还原和析氢描述符的金属间表面。该研究结合了机器学习技术和DFT计算,展示了预测电催化剂性能的可行性,实现了计算加速和成本节约。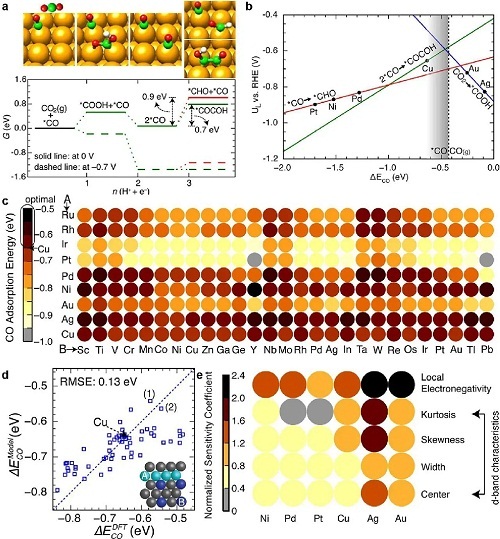
图3. 机器学习增强的化学吸附模型用于CO₂电还原催化剂筛选。(a) 在0 V和-0.7 V(相对于可逆氢电极)下,Cu(100)上CO₂电还原生成C1和C₂物种的最佳自由能路径。(b) CO₂电还原生成C1和C₂路径的理论极限电位作为反应性描述符(即CO吸附能)的函数。(c) 利用神经网络模型对第二代核-壳合金表面(Cu₃B-A@Cu单层)上的CO吸附能进行理性筛选。(d) 神经网络模型和DFT计算的特定Cu单层合金上CO吸附能对比。(e) 通过分析网络对输入特征扰动的响应获得的归一化灵敏度系数。
2.3 材料优化与设计
金/银纳米粒子(Au/AgNPs)在多个研究领域具有重大影响,其性质受尺寸、形态和表面化学性质的影响。为实现精确控制,需仔细考虑各种实验条件,这对可控合成构成了挑战。贝叶斯优化和遗传算法等机器学习算法可用于加速开发这一过程。例如,遗传算法基于光谱目标合成金纳米粒子,生成多种形态(图4a-4b)。在银纳米粒子制备中,微流控高通量实验平台与贝叶斯优化结合,高效探索参数空间(图4c)。引入深度神经网络(DNN)后,优化性能提升,吸收峰迅速向目标值收敛,预测光谱更加平滑。此外,机器学习方法还加速了TiO₂纳米管微图案和AgNPs掺杂的优化。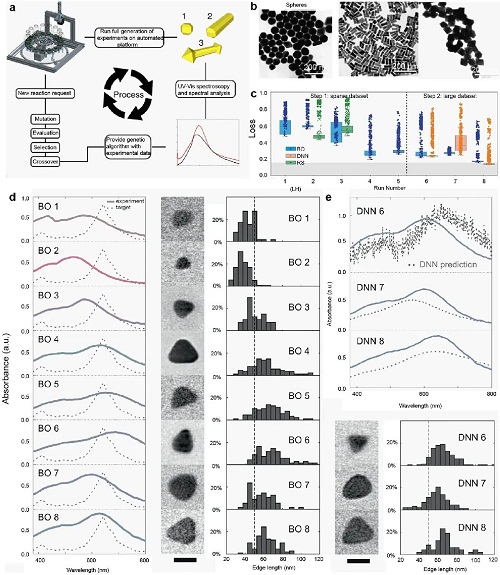
图4. 基于机器学习的金和银纳米粒子合成。(a) 平台工作流程的示意图,AuNPs的层级演化过程。(b) 金纳米球、金纳米棒和金纳米八面体的TEM图像。(c) 银纳米粒子合成过程的优化。(d) 最优贝叶斯优化(BO)模型和(e) DNN模型的吸收光谱,以及溶液中三角棱柱的同步尺寸分布分析。图像中的比例尺对应50纳米。
金属有机骨架(MOFs)是由金属节点和有机连接体组成的多孔材料,具有卓越孔隙率和巨大内表面积,在多种应用中具有广泛用途。利用决策树(DT)和支持向量机(SVM)模型,可高效识别高性能MOFs(图5a)。采用QSPR模型可在结构库中筛选具有潜力的甲烷纯化MOF材料(图5b)。图5c展示了MOFs特性交互散点图。DT基于简单规则预测,SVM减少计算需求。QSPR模型准确预测MOFs的CO₂工作容量和选择性,促进其在甲烷纯化中的应用,其中几何描述符如表面积和孔体积为关键特征。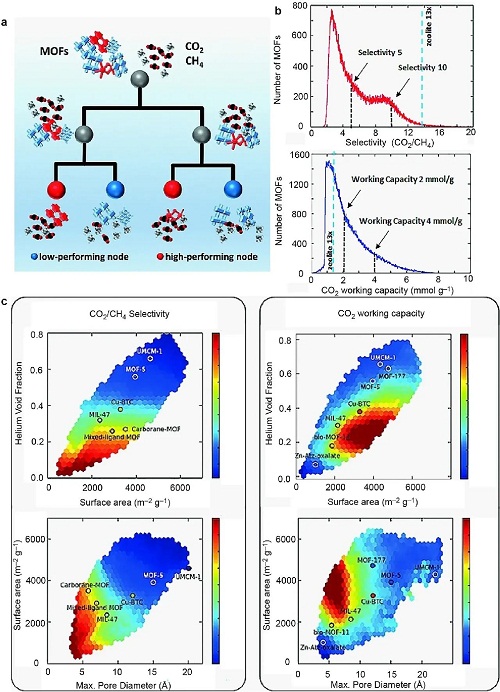
图5. 利用基于机器学习的定量结构-性质关系(QSPR)模型准确识别高性能金属-有机骨架材料:(a) 识别方法的示意图;(b) 筛选分子的数量作为单重态-三重态能隙(ΔEST)和振子强度的函数;(c) 基于时域密度泛函理论(TD-DFT)数据建立的线性模型预测与神经网络预测。
此外,通过巨正则蒙特卡洛(GCMC)模拟和综合数据库可用于探索材料结构与吸附性的关系,采用回归模型、径向分布函数等预测CO₂、N₂和CH₄的吸附量,并基于结构特性、不饱和度和电负性等化学特性预测MOFs中的吸附行为。图6a展示了决策树和泊松回归等算法,图6b-6e显示从DT到随机森林模型预测准确性的提升。结合结构和化学描述符的机器学习模型能高精度预测MOFs的甲烷吸附性能,有望加速MOFs筛选过程。但研究存在数据集较小的局限性,未来研究可使用更大、更多样化的数据集,并纳入其他气体吸附性质。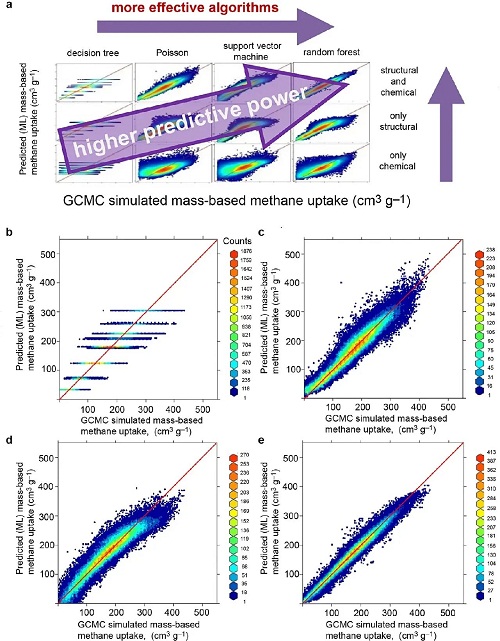
图6. 评估结构描述符和化学描述符以及包括决策树、泊松回归、支持向量机和随机森林在内的各种机器学习算法,以预测金属-有机骨架上的甲烷吸附量。(a) 引入了结构描述符和化学描述符,基于机器学习模型预测的甲烷吸附量(cm3 g⁻1)与通过GCMC模拟获得的结果比较,其中(b) DT、(c) 泊松(Poisson)、(d) SVM和(e) 随机森林(RF)模型均融入了结构和化学描述符。
2.4 过程模拟与调控
在移动机器人化学家领域,智能移动机器人通过自主实验加速了新材料的合成与优化,展现了AI在材料科学中的巨大潜力,但仍需降低构建成本和提高适应性。在生物医学方面,以机器学习为代表的AI技术被应用于双极电化学和微流控技术中,以优化生物材料的设计与制造过程,提高实验效率和准确性,为疾病诊断、治疗和检测等提供了新的可能性,推动生物医学的进步。
III 总结
大数据与计算能力的飞跃促进了人工智能,尤其是机器学习的迅猛发展,并在多个学科中展现广泛应用。在材料科学领域,机器学习虽已成功助力纳米材料制备,但仍处于起步阶段,面临着数据稀缺、模型可解释性不足、计算资源限制等难题。数据获取成本高昂且标准化处理缺失,而机器学习又高度依赖于高质量数据。为此,可解释AI技术和混合AI模型等新兴趋势正应运而生,力图克服这些障碍。此外,现有算法和模型通用性不足,且材料科学家对机器学习的了解尚浅。同时,机器学习与材料内在特性的一致性尚需深入验证。综上所述,该领域当前面临五大挑战:材料数据库的建设与管理、机器学习与高通量计算的融合、机器学习算法的改进、特征工程中的描述符选择与模型可解释性,以及材料数据格式和报告的标准化。展望未来,机器学习在材料科学中的应用潜力巨大,有望推动新材料研发与应用的持续增长,同时需兼顾伦理、可持续性和领域知识的深度融合。
作者简介

关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 斯坦福大学张兴才等:人工智能驱动的材料科学
 NML卷期 | 2025年第4期免费下载
NML卷期 | 2025年第4期免费下载 吉林师范大学鲁铭&东南大学应国兵等综述:MAX系列材料的多样性、合成、预测、性质及功能应用
吉林师范大学鲁铭&东南大学应国兵等综述:MAX系列材料的多样性、合成、预测、性质及功能应用 西交郭保林等:用于凝血障碍止血和糖尿病伤口愈合的多功能仿生冷冻凝胶
西交郭保林等:用于凝血障碍止血和糖尿病伤口愈合的多功能仿生冷冻凝胶 清华大学张萍、卢元等:“光虚拟芯片+人工智能”辅助的冠心病无创诊断
清华大学张萍、卢元等:“光虚拟芯片+人工智能”辅助的冠心病无创诊断