研究背景
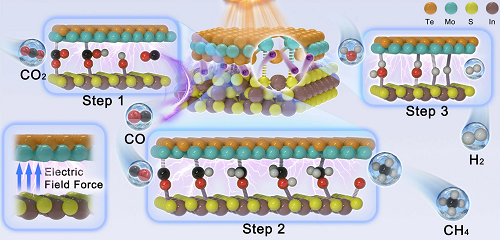
根据已有的研究,设计半导体纳米材料的双壳中空结构是提高光利用率、调节电子结构和化学键的空间相互作用、加速界面接触、提供更多催化反应位点和促进有效载流子分离和转移的最有效策略之一。鉴于此,本工作提出了Sᵥ-In₂S₃@2H-MoTe₃催化剂的“双壳纳米盒”设计,将2H-MoTe₃涂覆在Sᵥ-In₂S₃单壳纳米盒上,形成富含S-空位的Mo−S桥键结构。在强大的内置电场(IEF)和Sᵥ-In₂S₃@2H-MoTe₃(5)的Mo−S桥键的作用下,“S”-scheme电荷分离得到显著的提高,在380 nm计算出94.01%的内部量子效率。
Kangwang Wang, Zhuofeng Hu, Peifeng Yu, Alina M. Balu, Kuan Li, Longfu Li, Lingyong Zeng, Chao Zhang, Rafael Luque, Kai Yan*, Huixia Luo*
https://doi.org/10.1007/s40820-023-01221-3
本文亮点
1. S-空位导致Mo原子的d波段电子状态发生变化。
2. Mo−S桥键优化了吸附能量,加速了CO₂RR动力学。
3. 在380 nm波长下,光催化CO₂RR的内部量子效率为94.01%。
内容简介
受强界面电场的启发,中山大学严凯和罗惠霞教授等精心研制了具有强界面电场的Sᵥ-In₂S₃@2H-MoTe₃作为CO₂RR的高效催化剂。Sᵥ-In₂S₃@2H-MoTe₃(5)的中空双壳层异质结构扩大了比表面积,缩短了电荷转移距离,增强了传质能力,提高了光吸收能力,因而具有很强的光催化CO₂RR活性。更重要的是,S-空穴能促进CO₂的活化,降低形成*OCHO的能垒,促进电荷转移到*OCHO上,从而促进CO₂RR生成CO。最重要的是,丰富的界面异质结构形成了Mo−S桥键位点,导致了界面上的电荷差。电荷差导致在界面上形成Mo的极化位点,从而抑制了相邻中间产物的静电排斥,促进了CH₄的形成。这项工作可以通过光的利用、界面接触和反应位点,为开发用于大规模能源转换应用的优秀光催化剂开辟道路。
图文导读
I 结构表征
通过SEM、TEM、HRTEM、HAADF-STEM和HAADF-STEM-EDX元素图谱对Sᵥ-In₂S₃@2H-MoTe₃(5)、Sᵥ-In₂S₃、In₂S₃和2H-MoTe₃的形态特征进行了详细分析。如图1所示,Sᵥ-In₂S₃@2H-MoTe₃(5)的基本形貌是由大量超薄纳米片组成的双壳纳米盒,有利于活性表面的暴露和散射现象(Mie scattering)。Sᵥ-In₂S₃@2H-MoTe₃(5)的EDX元素映射图显示,Mo和Te元素明显分布在Sᵥ-In₂S₃的外表面(图1g),说明Sᵥ-In₂S₃与2H-MoTe₃之间成功形成了均匀的纳米异质结,进一步证明2H-MoTe₃是直接生长并附着在Sᵥ-In₂S₃上。此外,HRTEM图像、快速傅里叶反变换(IFFT)图和晶格条纹轮廓(LFP)显示出明显可见的晶格条纹,其间距为2.47和2.20 Å (图1e),分别精确指向In₂S₃的(219)晶面(JCPDS:73-1366)和2H-MoTe₃的(104)晶面(JCPDS:73-1650)。Sᵥ-In₂S₃@2H-MoTe₃(5)的选择面积电子衍射(SAED)图显示Sᵥ-In₂S₃和2H-MoTe₃呈环状,证实了复合材料存在多晶特征。

图1. Sᵥ-In₂S₃@2H-MoTe₃(5)的(a, b)SEM、(c, d)TEM、(e)HRTEM、IFFT和SAED、(f)HAADF STEM和(g)HAADF-STEM-EDS元素图谱。
II 电子结构分析
利用Mo K-edge和In K-edge的x射线吸收精细结构光谱(XAFS)深入研究了Sᵥ-In₂S₃@2H-MoTe₃中Sᵥ-In₂S₃和2H-MoTe₃(5)之间的原子和电子结构。在Mo和MoS₂中,Mo元素分别以0和+4氧化态存在。样品的吸收边缘介于Mo箔和MoS₂的吸收边缘之间。Sᵥ-In₂S₃@2H-MoTe₃(5)的Mo K-edge XANES光谱与MoS₂相比呈现负偏移。Sᵥ-In₂S₃@2H-MoTe₃(5)中Mo−S键的Mo价略有升高,与XPS结果一致。Sᵥ-In₂S₃@2H-MoTe₃(5)和Sᵥ-In₂S₃的In K-edge XANES光谱显示,Sᵥ-In₂S₃@2H-MoTe₃(5)和Sᵥ-In₂S₃中的In元素为+3价态(图2b)。In EXAFS光谱的优势峰位于1.67和1.77 Å,分别对应In−S和In−In配位(图2c)。如图2d和f所示,Mo位点的扩展x射线吸收精细结构(EXAFS)谱分别在1.99和2.85 Å处显示Mo – S和Mo -Mo键贡献的两个突出峰,这表明Sᵥ-In₂S₃@2H-MoTe₃中存在Mo−S桥接键(5)。此外,Sᵥ-In₂S₃@2H-MoTe₃(5)的Mo K-edge EXAFS光谱显示,与MoS₂相比,Mo−S键的径向距离正向移动(0.13 Å),进一步证实了Mo−S桥接键的存在。在EXAFS信号经小波变换(WT)后得到的二维图像中,Sᵥ-In₂S₃@2H-MoTe₃(5)(图2f)在8.5 Å处出现高能信号(红色区域),对应Mo−Mo配位键信号。
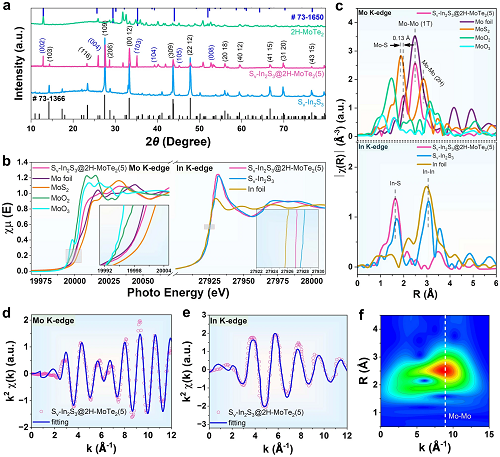
图2. 2H-MoTe₃、In₂S₃、Sᵥ-In₂S₃和Sᵥ-In₂S₃@2H-MoTe₃(5)的(a)XRD图谱;(b)Mo和In K-edge XANES光谱;(c)Mo和In在R空间的k3加权FT-EXAFS光谱。Sᵥ-In₂S₃@2H-MoTe₃(5)在k2加权k空间的(d)Mo K-edge EXAFS和(e)In K-edge EXAFS;(f)Sᵥ-In₂S₃@2H-MoTe₃(5)的k3加权EXAFS等高线图。
III 光催化CO₂RR性能
图3a给出了2H-MoTe₃、Sᵥ-In₂S₃和Sᵥ-In₂S₃@2H-MoTe₃(5)的CO₂-TPD图,揭示了在所研究的温度范围内存在显著的峰,从而表明光催化剂表面存在适度的初级CO₂吸附中心。Sᵥ-In₂S₃@2H-MoTe₃(5)的中空多孔特性具有较高的CO₂捕获能力(36.83 cm3·g⁻1,图3b),促进了Sᵥ-In₂S₃@2H-MoTe₃(5)上的CO₂RR。Sᵥ-In₂S₃@2H-MoTe₃(5)、In₂S₃和Sᵥ-In₂S₃表现出相对较低的光催化CO₂RR活性,CH₄演化率分别为13.97、1.53和2.32 μL·h⁻1(图3d和e),而2H-MoTe₃几乎不能光催化CO₂RR,与上述表征完全一致。特别是Sᵥ-In₂S₃@2H-MoTe₃(5)的CO₂-to-CH₄转化率高达70% (CH₄选择性为79.6%),在420 nm处的最佳表观量子效率(AQE)值为16.5%。如图3g所示,使用Sᵥ-In₂S₃@2H-MoTe₃(5)进行6个循环反应后,CH₄、CO和H₂的析出量没有明显减少。

图3. Sᵥ-In₂S₃、2H-MoTe₃和Sᵥ-In₂S₃@2H-MoTe₃(5)的(a)CO₂-TPD图谱。样品在298 ℃下的(b)CO₂吸附等温线;(c)几种条件下的对照实验;(d)CO₂转化率和产物选择性;(e)CO、CH₄和H₂的产率;(f)Sᵥ-In₂S₃@2H-MoTe₃(5)的AQE, IQEcr和吸收光谱;(g)Sᵥ-In₂S₃@2H-MoTe₃(5)的稳定性测试。
IV 光电性能分析
我们通过稳态PL、超快飞秒瞬态吸收(fs-TA)光谱和TRPL光谱来评估载流子的重组,以研究光催化CO₂RR的活性。显然,2H-MoTe₃表现出最强的发射峰,对应于电子-空穴对的快速重组(图4a),表明Sᵥ-In₂S₃@2H-MoTe₃(5)的电子导电性增强。其中,Sᵥ-In₂S₃@2H-MoTe₃(5)为最弱的发射峰,与它们的最佳光催化CO₂RR性能一致。此外,我们还进行了表面光电压(SPV)光谱来验证其载流子转移机理,如图4f所示。结果表明,2H-MoTe₃在整个波长内不存在SPV信号,说明其光载流子分离效率较差。这就是为什么2H-MoTe₃表现出极差的CO₂RR活性。相比而言,在In₂S₃和Sᵥ-In₂S₃的SPV光谱中可以观察到明显的正光电压响应,说明空穴向In₂S₃和Sᵥ-In₂S₃表面迁移,这是n型半导体的典型特征。此外,在SPV光谱中,与2H-MoTe₃和Sᵥ-In₂S₃的正光电压信号不同,Sᵥ-In₂S₃@2H-MoTe₃(5)在300 ~ 430 nm处出现了一个显著增强的负光电压信号,说明Sᵥ-In₂S₃的光生电子和2H-MoTe₃的空穴分别转移到光照侧和背光侧,2H-MoTe₃的剩余电子通过内置电场与Sᵥ-In₂S₃的空穴复合。这进一步揭示了异质结内部通过“S”-scheme途径有效的界面电荷转移。

图4. (a)样品的PL光谱;(b)Sᵥ-In₂S₃、(c)In₂S₃、(d)2H-MoTe₃和(e)Sᵥ-In₂S₃@2H-MoTe₃(5)在不同时间下的Fs-TA光谱;(f)样品的SPV光谱;(g)Sᵥ-In₂S₃和2H-MoTe₃的归一化激子漂白信号衰减(曲线为公式S1和S2拟合结果);(h)Sᵥ-In₂S₃@2H-MoTe₃(5)的电荷密度差图(品红色区域(正值)和青色区域(负值)分别表示电子的积累和消耗);(i)IQEpc作为光照光子能量的函数;(j)IQEpc作为样本的带隙倍数(hυ/Eg)的函数。
V Mo−S桥接与CO₂RR活性的关系
利用原位漫反射-红外傅里叶变换光谱(DRIFTS)和原位高分辨率XPS光谱分析了表面特征与光催化CO₂RR之间的关系。分析结果如图5所示。从0 ~ 60 min,随着光照时间的增加,出现新的吸收峰,其强度逐渐增大。在1127 cm⁻1处观察到的红外峰,可归因于CH₃O* (星号表示催化活性位点),而在1040 cm⁻1处的峰可归因于CHO*的特征波段。1560和1630 cm⁻1处的吸收峰属于COOH*,COOH*通常被认为是CO₂RR为CH₄或CO以及CH₃OH的关键中间体。1430 cm⁻1处的峰对应于HCO₃*的对称拉伸。单齿碳酸盐(m-CO₃2⁻)和双齿碳酸盐(b-CO₃2⁻)的形成分别来自1368和1329 cm⁻1左右的红外吸收峰(图5a)。Sᵥ-In₂S₃也有类似的现象(图5b)。利用原位XPS研究了光催化CO₂RR过程中Sᵥ-In₂S₃@2H-MoTe₃(5)表面碳氢化合物的变化。在暗态下,没有观察到气相CH₄ (286.9 eV)的峰值(图5c和d),说明CO₂RR是光驱动的。相反,随着光照的逐渐增加,表面CHₓ物种在285.8 eV左右出现峰值,进一步支持催化剂表面生成的CH₄解离生成H₂,这也是我们在混合产物中检测到H₂产物的原因。
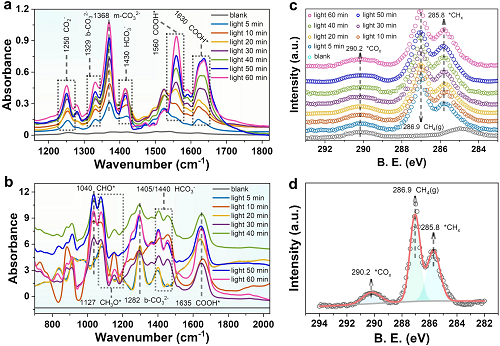
图5. 光催化CO₂RR在(a)Sᵥ-In₂S₃@2H-MoTe₃(5)和(b)Sᵥ-In₂S₃上的原位红外光谱;(c)Sᵥ-In₂S₃@2H-MoTe₃(5)在不同光照时间下的原位C1s XPS光谱;(d)在光照5min下通过Sᵥ-In₂S₃@2H-MoTe₃(5)收集CH₄转化的C1s XPS光谱。
基于上述分析,我们进行了DFT模拟,以深入了解Sᵥ-In₂S₃@2H-MoTe₃(5)上CO和CH₄产物的Mo−S桥接键机制。在本研究中,为了更准确地解释CO₂RR为CO、H₂和CH₄的热力学过程,我们在可见光照射下将S-空位引入到In₂S₃的结构中,确定S-空位参与光催化反应的最佳位置。最初,CO₂会逐渐吸附在Sᵥ-In₂S₃@2H-MoTe₃(5)表面,同时溶液中的H₂O会被解离,生成OH−和H+。值得注意的是,路径1在反应过程中是吸热的(ΔG > 0),因此这里不考虑它。CH₄自由能图如图6c所示,对应的最小能量反应途径如图6d所示。自由能计算图表明,*OCHO与CO和*OH的反应过程是一个潜在的决定性步骤 (ΔG = − 0.84 eV)。最初,CO₂在能量上有利于从CO₂到CO的Mo−S桥接键位点。当一个H原子靠近吸附的CO₂时,它可以形成*OCHO。值得注意的是,S-空位可以促进CO₂的活化,降低*OCHO形成的能垒,促进电荷向*OCHO转移,从而促进CO₂RR形成CO。此外,*OCHO的形成是最终CH₄形成过程中具有最高能垒的步骤,因此,*OCHO将跃迁到OH*。CO*的解吸的ΔG比CHO*解吸的ΔG低−0.51 eV(图6c),导致CO₂RR过程中最终产物在Mo−S桥接键位点与CO、H₂和CH₄混合。需要强调的是,Sᵥ-In₂S₃@2H-MoTe₃(5)上CO的解吸是一个放热过程,而CO*加氢成CHO*是自发放热的,即ΔG < 0,这使得CO₂RR成CO的选择性更好,这与上文CO₂-TPD的结果分析非常吻合(图3a)。
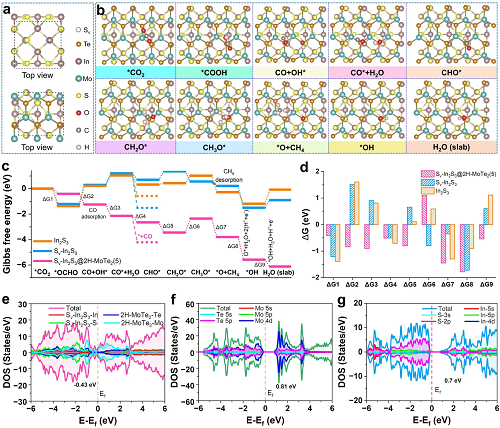
图6. (a)理论计算中Sᵥ-In₂S₃@2H-MoTe₃(5)的原子模型;(b)Sᵥ-In₂S₃@2H-MoTe₃(5)界面上CO₂RR过程的吸附原子结构示意图;(c)Sᵥ-In₂S₃@2H-MoTe₃(5)、Sᵥ-In₂S₃和In₂S₃的CO₂RR的吉布斯能量分布图和(d)能量变化图;(e)Sᵥ-In₂S₃@2H-MoTe₃(5)、(f)2H-MoTe₃和(g)Sᵥ-In₂S₃的态密度。
作者简介
 罗惠霞
罗惠霞本文通讯作者
 严凯
严凯本文通讯作者
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 中山大学罗惠霞、严凯等:理解光催化CO₂还原的桥接位点和加速量子效率
 西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化
西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化