Yangwu Chen, Dingtao Ma*, Kefeng Ouyang, Ming Yang, Sicheng Shen,Yanyi Wang, Hongwei Mi, Lingna Sun, Chuanxin He, Peixin Zhang*
Nano-Micro Letters (2022)14: 154
https://doi.org/10.1007/s40820-022-00907-4
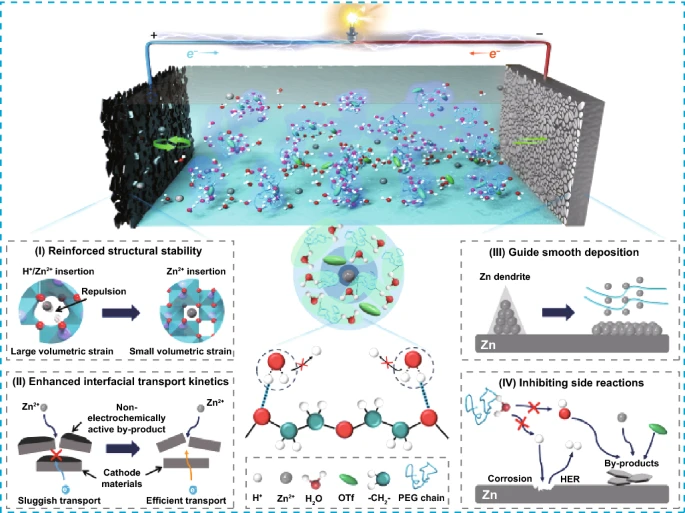
1. 在水性电解液中引入 PEG 400 添加剂能够调节 Zn²⁺ 溶剂化结构并抑制游离水分子的电离。
2. 这种反质子电解液不仅可以减少正极宿主材料的晶格膨胀,抑制相关副产物,还可以引导Zn均匀沉积,抑制析氢反应。
3.通过一体化协同作用机制可以实现具有18,000次循环寿命的高倍率 Zn-V₂O₃/C 电池。
分子动力学模拟结果表明,在0PEG电解液中Zn²⁺主要与2个 OTf ⁻ 和 4个 H₂O配位,并有大量的自由水分子存在。引入PEG400分子后,在50PEG电解液中Zn²⁺主要与4个 OTf ⁻ 和 2个 H₂O配位,拥有很少的自由水分子,并且,PEG分子链能够打破水分子间的氢键网络,从而抑制水分子的电离。因此50PEG比0PEG电解质拥有更宽的电化学稳定窗口和底的腐蚀电位。
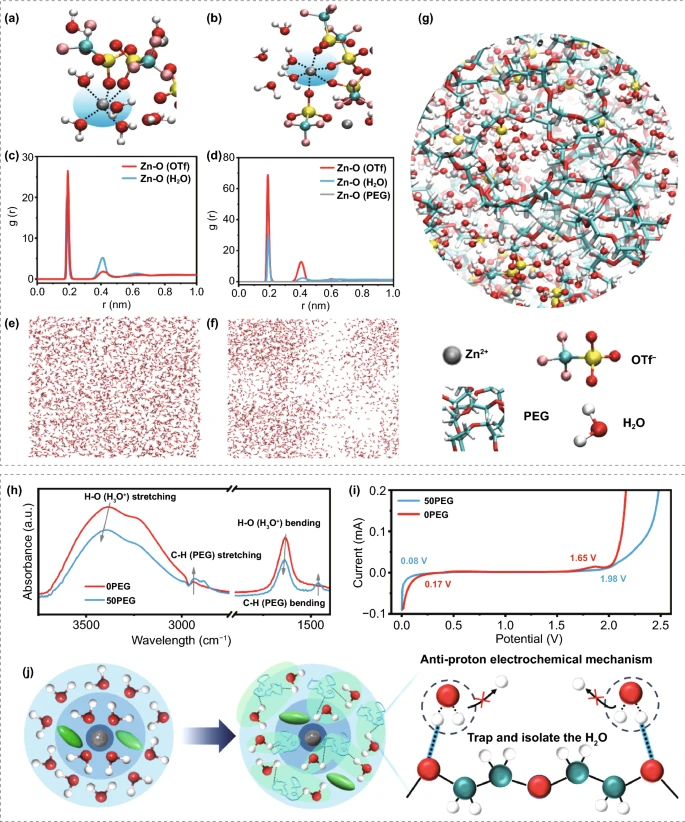
图1. (a)0PEG和(b)50PEG 电解液中Zn²⁺的溶剂化结构;(c)0PEG和(d)50PEG 电解液中Zn²⁺的径向分布函数RDFg(r);(e)0PEG和(f)50PEG 电解液中仅含水分子的模拟快照图;(g) 50PEG 电解液中富含 PEG 的区域吸收水分子的快照图;(h)0PEG 和 50PEG 电解液标准化的FTIR 光谱;(i)0PEG和50PEG 电解质在0.1 mV s⁻¹ 扫描速率下的LSV 曲线;(j)PEG添加剂调控的反质子电化学机制。
为了揭示这种反质子电解液对V₂O₃/C正极储锌能力的影响,组装了Zn-V₂O₃/C电池,并对其进行了电化学性能测试。图3(a-c)表明电解液改性后倍率性能有了明显的提升,50PEG比0PEG电解液能实现优异的倍率性能和容量保持率以及更低的极化。同时,这种储锌能力也优于先前报道的许多其他类型的正极材料。
在5A g⁻¹电流密度下,尽管在0PEG电解液中的电极能实现比50PEG电解液更高的比容量,但在0PGE电解液中V₂O₃/C电极经历活化后,容量急速衰减,变现出极差的循环稳定性。相反,在50PEG电解液中V₂O₃/C电极能稳定循环6000圈后容量还有222.5 mAh g⁻¹。即使在20A g⁻¹的电流密度下循环18000次循环后,仍能实现优异的循环稳定性,保留容量为121.8mAh g⁻¹,库伦效率接近100%,这样的超稳定循环性能也明显高于以往的很多报道。
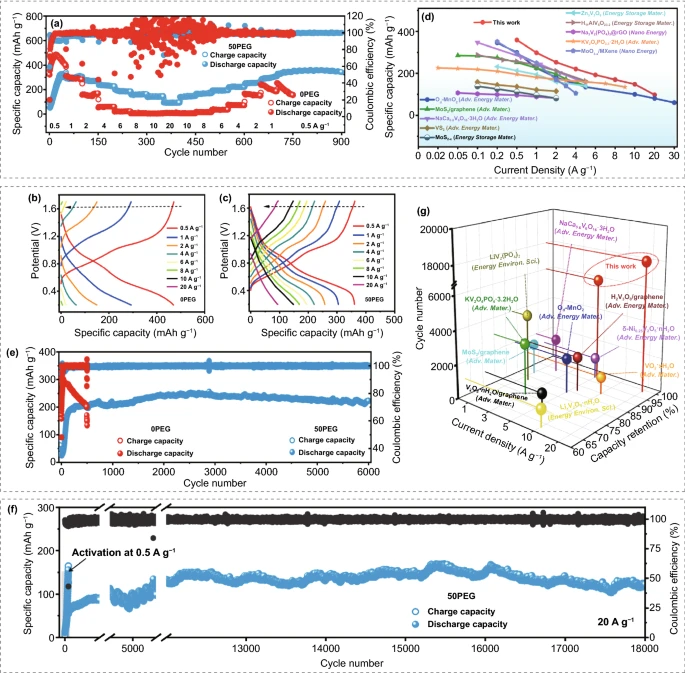
图3. (a)V₂O₃/C正极的倍率性能测试;(b,c) V₂O₃/C正极在不同电流密度下相应的充放电曲线;(d)本工作与前人工作的倍率性能的比较;(e)V₂O₃/C正极在5A g⁻¹和(f)20A g⁻¹电流密度下进行循环稳定性测试;(g)本工作与其他报告的循环稳定性的比较。
IV 水系Zn-V₂O₃/C电池的储锌机制
为了研究V₂O₃/C电极的存储机理,并进一步揭示这种反质子电解液对Zn²⁺存储行为的影响,利用原位电化学拉曼光谱初步研究了电极的结构演变。如图4(a-b)所示,在首圈的充电过程中,所有振动峰的强度都逐渐减弱和变宽。透射电子显微镜和相应的HRTEM图像(图4C)显示了V₂O₃在初始充电到1.7V后的晶格畸变。此外,还研究了V₂O₃/C电极首圈循环过程中V 2p和Zn 2p的非原位XPS谱,如图4E所示,当充电到1.7V时,V³⁺/V²⁺的比值将从1.9(初始状态)增加到4.43,但是,这个值在随后放电到0.2V后才会下降到3.81。这一结果表明V₂O₃的晶格畸变是不可逆的。而在高分辨率的锌2p XPS谱(图4f)中,在初始态没有检测到锌的信号峰,然而,在放电到0.2V后出现了Zn 2p2/3和Zn 2p1/2信号峰,这主要归因于Zn²⁺嵌入到畸变的V₂O₃晶格中。
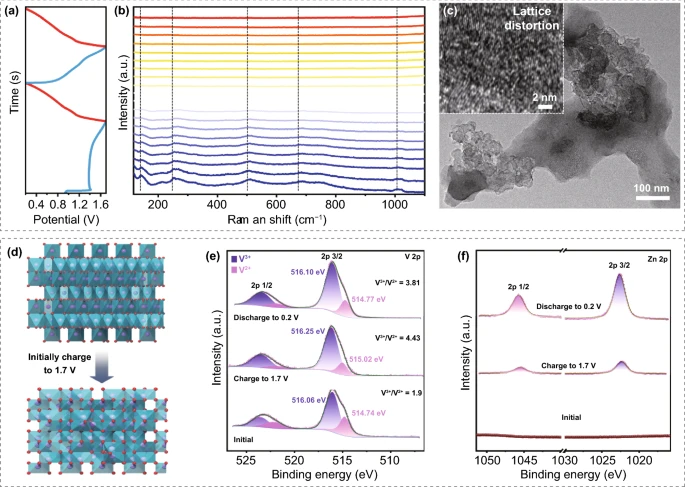
V V₂O₃/C电极的储能机理研究
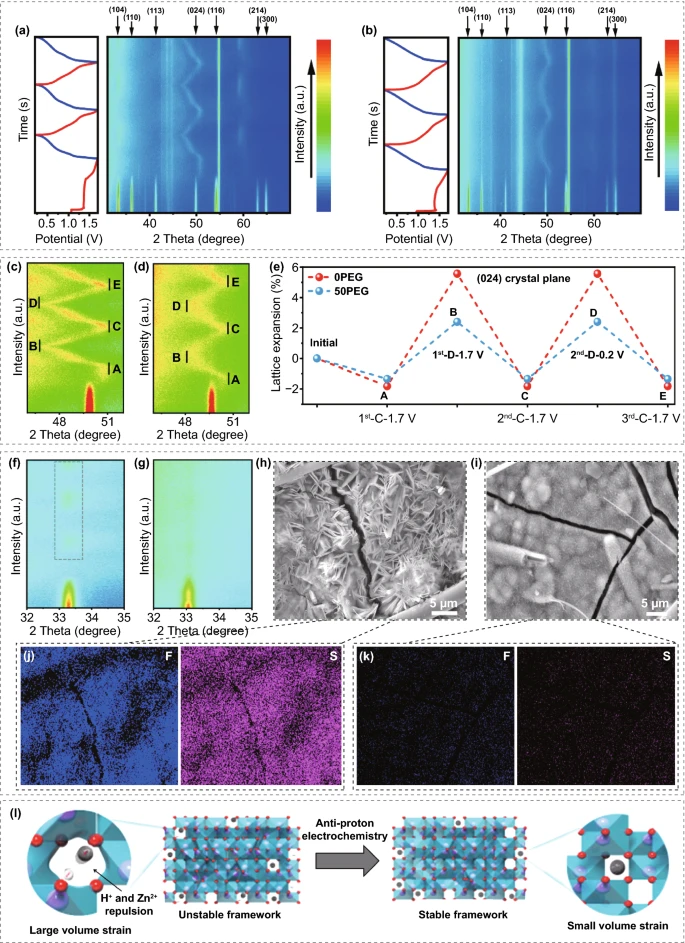
采用原位X射线衍射进一步研究其储锌机理。在首圈充电过程中,V₂O₃的衍射峰逐渐减弱,直到充电到1.7V,衍射峰完全消失。在两种电解液中,只有(024)面向右移动(图5c-d中的A点),结合上述原位拉曼和高分辨透射电子显微镜的表征,这一过程归因于V₂O₃的晶格畸变。随着后续放电过程的继续,(024)晶面将逐渐向左点(B)移动,对应于Zn²⁺嵌入引起的晶格膨胀,此外,在接下来的两个循环中,Zn²⁺的嵌入/脱出具有极高的可逆性,表明畸变后的V₂O₃能高效的储存Zn²⁺。0PEG电解液中的晶格膨胀率远大于50PEG电解液中的晶格膨胀率,此外,在0PEG电解液体系(图5f)中能观察到33.31°处的杂质(Znₓ(OTf)ᵧ(OH)₂ₓ₋ᵧ·nH₂O)峰,这种现象归因于H⁺和Zn共嵌入到宿主材料导致过度的晶格膨胀以及局部pH升高导致生成碱式副产物。
此外,V₂O₃/C电极以0.5A g⁻¹电流密度在0PEG电解液中循环200圈后的电极表面形貌存在大量片状附着物,相应的能谱图显示出强烈的F,S信号,进一步确认了Znₓ(OTf)ᵧ(OH)₂ₓ₋ᵧ·nH₂O的生成。与之相反,在50PEG电解液中循环后V₂O₃/C电极表面没有明显的片状副产物形成。因此,在50PEG电解液中可以显著抑制H⁺的嵌入行为。在反质子电解液的作用下,V₂O₃/C电极在酸性电解液中的Zn²⁺、H⁺共嵌入机理可以调节为以Zn²⁺为主的嵌入机理。事实上,虽然双离子插入可以带来更高的比容量,但质子嵌入的抑制可以避免H⁺和Zn²⁺之间的大的静电斥力,这通常会导致大的晶格膨胀,破坏主体骨架。
VI 水系Zn-V₂O₃/C电池的传输动力学研究
在0PEG电解液中,由于正极-电解质界面副产物的形成会极大地影响离子传输和电荷转移,因此,0PEG电解质体系中的界面转移阻抗Rct随着循环的进行呈现逐渐增加的趋势,这不仅消耗了电解质的 Zn²⁺,而且阻碍了循环过程中离子/电子的有效传输。与此形成鲜明对比的是,在50PEG电解质体系中,在最初的两个循环中界面转移阻抗Rct仅略有增加,并且在随后的循环中仍能保持稳定,代表了其高效稳定的界面传输动力学。
V₂O₃/C电极在0PEG电解液中的GITT曲线表明,电池的比容量在充放电过程中不断下降,甚至出现过压区,如阴影区所示。这种过电压区域通常代表缓慢的离子扩散,这可能是由于H⁺的自发预嵌入或 H⁺/Zn²⁺ 的竞争性嵌入,以及正极-电解质界面中副产物的产生。不同的是,50PEG 电解质中的电极显示出稳定的比容量,没有任何过电压区域。原位 EIS 和 GITT 研究的结果表明副产物的形成会导致界面离子/电子传输缓慢,这种不利影响将进一步降低活性材料的利用率,从而大大削弱电极的倍率性能和循环稳定性。
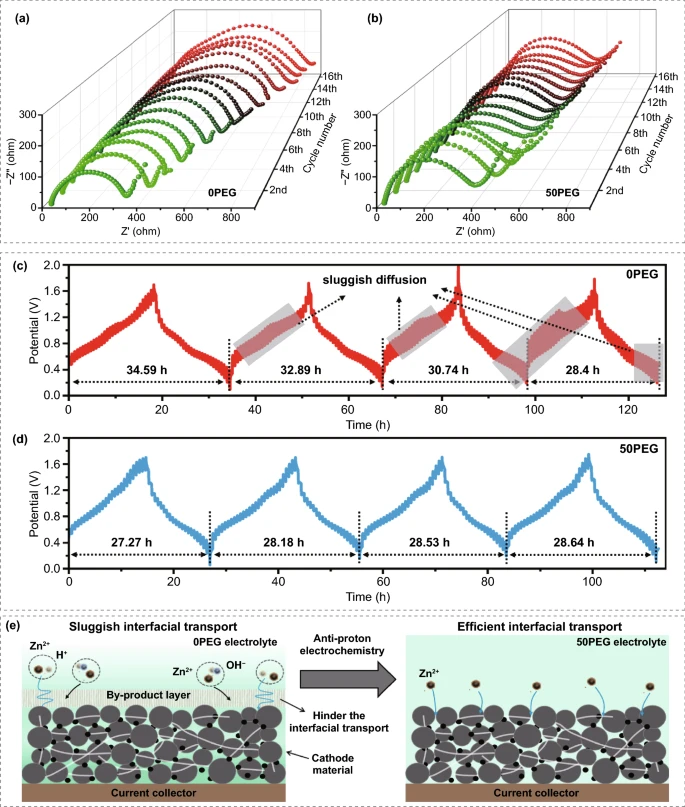
图6. 在0.5A g⁻¹的电流密度下,V₂O₃/C电极在(a)0PEG和(b)50PEG电解液中的前16次循环的原位电化学交流阻抗谱;脉冲电流密度为 0.2 A g⁻¹,脉冲时间为 5 分钟,弛豫时间为 30 分钟,V₂O₃/C 电极的在(c)0PEG和(d)50PEG 电解液中恒电流间歇滴定技术测试;(e)由反质子电解液调节的电极界面传输动力学的演变。
VII Zn||Zn 对称电池的电化学性能
由于锌金属负极在影响 AZIBs 的电化学性能方面也起着关键作用,因此还研究了这种反质子电解液对调节 Zn 电镀/剥离行为和 HER 现象的影响。在 0PEG 电解液中,可以清楚地观察到锌金属负极表面枝晶的快速生长,相反,即使在 20 分钟后,在 50PEG 电解质中也可以实现光滑而致密的沉积表面,该结果表明PEG分子链有利于引导Zn均匀沉积。Zn||Zn 对称电池的循环结果也表明在0PEG 电解液中的对称电池快速短路,而在50 PEG 电解液中锌电镀/剥离的可逆性可以显着增强。同时在0PEG电解液中循环200小时后电池厚度呈现明显的膨胀,这归因于析氢反应(HER),而在50PEG电解液中循环后的电池没有明显的膨胀。
同时,相应的GF隔膜的数码照片和循环后的Zn金属负极的FESEM图谱进一步证明了0PEG电解液中的锌枝晶生长甚至刺穿隔膜,在 50PEG 电解液条件下可以观察到平滑的锌沉积,没有明显的枝晶形成。对于锌金属负极侧,我们证明了这种反质子电解液不仅有利于引导锌的均匀沉积,而且有利于抑制 HER 和副产物的产生。
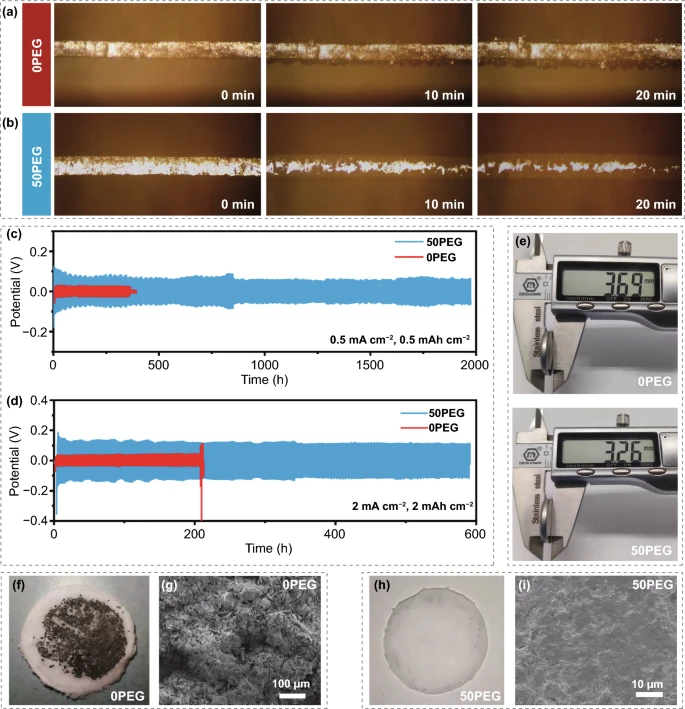
图7. 在 10 mA cm⁻² 电流密度下原位光学观察 (a)0PEG 和(b)50PEG 电解液中的 Zn 沉积;Zn||Zn对称电池在(c) 0.5 mA cm⁻²、0.5 mAh cm⁻²和(d) 2 mA cm⁻²、2 mAh cm⁻²电流密度下的循环稳定性; (e) Zn||Zn 对称电池在 2 mA cm⁻² 和 2 mAh cm⁻²电流密度下循环 200 h 后的厚度,以及相应 GF 隔膜的数码照片和相应 Zn 金属负极的 FESEM 图像(f,g) 0PEG 和 (h,i) 50PEG 电解质。

本文通讯作者
2、超级电容器。
▍主要研究成果
▍Email:pxzhang@szu.edu.cn

Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 多功能反质子电解液:可用于高倍率和超稳定水系锌-钒氧化物电池
 西交利物浦大学赵春/刘庆&华师大田博博等综述:突触可塑性工程从器件到系统
西交利物浦大学赵春/刘庆&华师大田博博等综述:突触可塑性工程从器件到系统 湖北大学梅涛&香港城市大学郭再萍院士等:两性离子COF调控 Li⁺传输与界面稳定性实现高性能锂金属电池
湖北大学梅涛&香港城市大学郭再萍院士等:两性离子COF调控 Li⁺传输与界面稳定性实现高性能锂金属电池 东南大学孙正明/张培根等: 一维金属异质结构磁-介电协同增强低频微波吸收奏
东南大学孙正明/张培根等: 一维金属异质结构磁-介电协同增强低频微波吸收奏 北科大王存海/哈工大王富强等综述: 动态辐射制冷—智能热管理的机制、策略和应用
北科大王存海/哈工大王富强等综述: 动态辐射制冷—智能热管理的机制、策略和应用