研究背景
鉴于能源需求日益增长,开发高能量密度、低成本的可充电锂电池愈发引起人们的重视。一氧化硅阳极由于其高容量特性和低成本优势具有巨大的应用前景。为了缓解一氧化硅阳极在脱嵌锂过程中的体积膨胀效应,发展了材料结构设计、表面涂层修饰和元素掺杂改性等一系列技术方法。但电解液与一氧化硅阳极的固液界面稳定机制往往被人忽视,因此深入开发与一氧化硅阳极高兼容性和高稳定性的新型电解液,进而解析锂离子溶剂化学对于锂离子脱溶剂化/溶剂化过程及界面演变行为的影响机制对于开发长寿命硅基锂离子电池具有重要意义。

Breaking solvation dominance effect enabled by ion-dipole interaction toward long-spanlife silicon oxide anodesShengwei Dong, Lingfeng Shi, Shenglu Geng, Yanbin Ning, Cong Kang, Yan Zhang, Ziwei Li, Jiaming Zhu, Zhuomin Qiang, Lin Zhou, Geping Yin, Dalong Li*, Tiansheng Mu*, Shuaifeng Lou*
Nano-Micro Letters (2025)17: 95
https://doi.org/10.1007/s40820-024-01592-1
本文亮点
1. 基于塑性晶体丁二腈的深共晶电解液,内部存在强烈的离子-偶极相互作用,从而构筑了富阴离子的锂离子溶剂化结构,同时保持了高离子导电性和锂离子迁移数。
2. 从分子水平精确调控多种离子-离子、离子-偶极和偶极-偶极相互作用,有助于锂离子溶剂化结构从溶剂分子主导转变为阴离子主导,从而稳定一氧化硅阳极界面。
3. 通过光学显微镜和Micro-CT分析可以证明,阴离子衍生的SEI能够有效缓解一氧化硅阳极脱嵌锂过程中的不可逆体积膨胀。
内容简介
一氧化硅阳极与电解液的界面稳定与演变机制对于硅基锂离子电池电化学性能的表达至关重要。在本研究中,哈尔滨工业大学娄帅锋等人提出了一种由丁二腈(SN)和双三氟甲基磺酰亚胺锂(LiTFSI)组成的新型深共晶电解液。并通过引入弱溶剂化能力的氟代碳酸乙烯酯溶剂以调节Li⁺与SN之间的离子-偶极相互作用。该电解液表现出高比例的接触离子对(CIP)、阴离子聚集体(AGGs)和低比例的溶剂化离子对(SSIP)。在ST-F电解液中,富含FEC和阴离子的独特Li⁺溶剂化结构选择性地在一氧化硅阳极上分解,进而形成富含LiF的稳定SEI。该SEI增强了固液界面的稳定性和电化学性能,有效地保护阳极免受体积膨胀和电解液持续分解引起的性能衰减。因此,ST-F电解液的独特离子溶剂化结构以及高稳定性富无机物界面设计有望推动硅基阳极在高能量密度电池中的应用。
图文导读
I 丁二腈电解液溶剂化学
图1a展示了丁二腈基深共晶电解液形成原理,由于C≡N键强极性和电负性,SN溶剂分子进入锂离子溶剂化鞘层,从而形成稳定的电解液体系。由于SN的良好电化学稳定性,SN基电解质的电化学窗口扩展到5.0 V,远超传统碳酸酯电解液(图1b)。ST电解液和ST-F电解液展示了室温下的电导率分别为2.08 mS cm⁻¹和2.78 mS cm⁻¹(图1c),以及锂离子迁移数分别为0.21和0.30(图1d)。在锂离子溶剂化鞘层中,存在各种处于竞争关系的相互作用力,包括离子-离子相互作用、离子-偶极相互作用和偶极-偶极相互作用。ST和LE电解液中强烈的Li⁺-偶极相互作用导致TFSI⁻减少,弱溶剂化能力的FEC的引入可减弱离子-偶极相互作用,促进了Li⁺和TFSI⁻之间的离子-离子作用,从而导致大量游离TFSI⁻进入锂离子溶剂化鞘层,形成富含阴离子的独特溶剂化结构(图1e)。
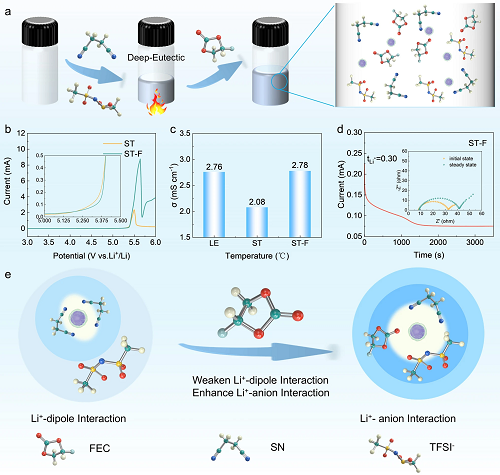
图1. 丁二腈基电解液的制备及电化学性质测试:(a)丁二腈基深共晶电解液形成原理示意图;电化学性质测试:(b)电化学窗口测试;(c)室温下离子电导率测试;(d)锂离子迁移数测试;(e)FEC调控溶剂化结构作用机制图。
结合密度泛函理论(DFT)计算解析丁二腈基电解液的特征溶剂化结构。从分子静电势分布可以看出,SN中的氮原子和FEC中的氧原子表现出与Li⁺强烈的配位作用。图2a展示了丁二腈电解液中各成分的LUMO和HOMO能级。相对较低的EC(-1.61 eV)、LiTFSI(-1.09 eV)和FEC(-0.33 eV)的LUMO能级,SN(0.73 eV)相比更高的还原可能性,此外SN的较低HOMO能级(-9.43 eV)表明其具有显著的抗氧化特性和应用于高电压电池的巨大潜力。如图2b所示,Li⁺-FEC的结合能低于Li⁺-EC和Li⁺-SN,表明FEC的溶剂化能力较弱。图2c描述了ST-F电解液中FEC对于溶剂化结构的调节作用。结合分子动力学模拟(MD)进一步分析了ST和ST-F电解液中锂离子溶剂化结构,如图2d~f所示,在ST电解液中,Li⁺-N(SN)的配位数为1.61,而Li⁺-O(TFSI⁻)的配位数为0.48。在ST-F电解液中, Li⁺-O(TFSI⁻)的配位数明显增加至0.74,表明FEC减弱了Li⁺溶剂化结构中的离子-偶极(Li⁺-N)相互作用,并增强了离子-离子相互作用(Li⁺-TFSI⁻)。由于Li⁺-FEC的离子-偶极相互作用较弱,ST-F电解液中Li⁺-TFSI⁻相互作用增强,导致阴离子更多地参与Li⁺溶剂化结构。此外,图2g表明AGGs和CIP的比例分别为12.7%和58.7%,高于ST电解液中的比例(10%和30.6%)。
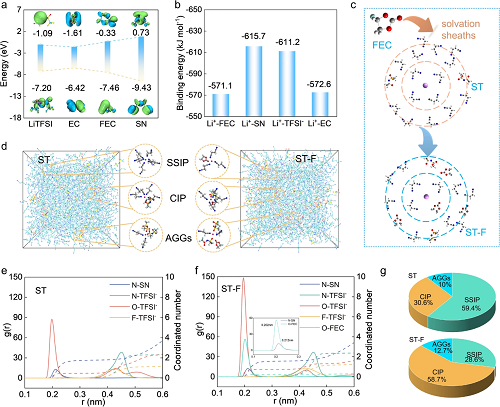
图2. 丁二腈基电解液的DFT计算和溶剂化结构分析:(a)LUMO和HOMO能级;(b)结合能计算;(c)FEC调控丁二腈基电解液溶剂化结构示意图;(d)ST和ST-F电解液的MD模拟快照。不同电解液的RDF:(e)ST电解液;(f)ST-F电解液;(g)比较丁二腈基电解液SSIP、CIP和AGGs的比例。
利用傅立叶变换红外光谱和拉曼光谱解析丁二腈电解液中独特Li⁺溶剂化结构。红外光谱显示了大约2950 cm⁻¹和2253 cm⁻¹处两个典型峰,分别对应于SN中的C-H拉伸振动和C≡N的拉伸振动(图3a)。在拉曼光谱中,721 cm⁻¹、729 cm⁻¹和741 cm⁻¹处的特征峰分别被归属为游离TFSI⁻、CIP和AGGs(图3b)。与ST电解液相比,ST-F电解液中AGGs的比例较高。结合⁷Li液体核磁共振波谱以深入阐明Li⁺的溶剂化环境(图3c)。引入FEC导致共振信号向-0.80 ppm的下移,表明Li⁺周围的阴离子浓度增加。图3d展示了SSIP、CIP和AGGs的典型溶剂化结构。图3e展示了不同温度下的ST-F电解液离子电导率显著高于ST电解液。与LE和ST电解液相比,ST-F电解液表现出较低的扩散势垒(0.034 eV)和优越的Li⁺扩散动力学(图3f)。图3g展示了丁二腈电解液中的Li⁺脱溶化过程。由于FEC具有弱溶剂化能力和较低的脱溶化障碍,因此展现出优越的Li⁺扩散动力学。
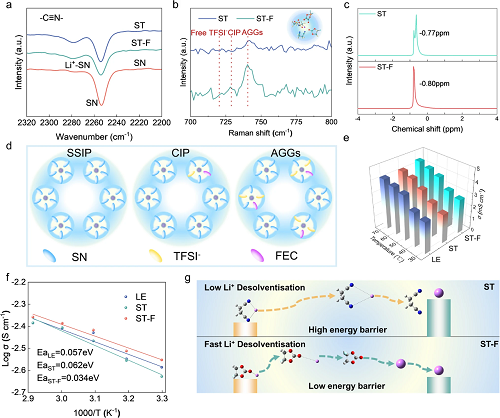
图3. 锂离子溶剂化结构和迁移动力学分析:(a)傅里叶变换红外光谱测试;(b)拉曼光谱测试;(c)⁷Li核磁共振波谱测试;(d)SSIP、CIP和AGGs结构示意图;(e)不同温度下的离子电导率测试;(f)电解液活化能计算;(g)ST和ST-F电解液中锂离子脱溶剂化过程示意图。
II 界面演变与电化学性能测试
循环伏安测试揭示SiO阳极的合金化/去合金化行为。ST-F电解液中的1.42 V处的分解峰可被归因于LiTFSI和FEC的协同分解效应(图4a)。通过不同扫描速率下的循环伏安曲线测试,可以发现ST-F电解液和ST-F电解液中的SiO阳极表现出优异的合金化/去合金化动力学(图4c、b)。在图4d中,SiO阳极在ST-F电解液中表现出优异的循环稳定性,可归因于丰富的FEC/阴离子溶剂结构的协同还原和分解,形成了稳定的SEI。在图4e中,SiO阳极在ST-F电解液中展现出良好的倍率性能,在2 A g⁻¹的电流密度下仍保持822.1 mAh g⁻¹的比容量。相比之下,ST电解液展示了596.1 mAh g⁻¹的剩余容量,LE电解液展示了502.2 mAh g⁻¹的剩余容量(图4f)。如图4g所示,在ST电解液中SiO阳极展现出严重的副反应,其阳极表面SEI厚度约为50 nm。相比之下,ST-F电解液中的富阴离子溶剂化结构促进了薄而致密SEI的生成(30 nm)。同LiCoO₂在ST-F电解液中展示出优异的高电压性能,这是由于C≡N与过渡金属离子之间的稳定络合作用保证了LiCoO₂正极的稳定循环(图4h)。因此,使用ST-F电解液组装的SiO|LiCoO₂全电池展现出优异的循环稳定性。这表明通过调整离子-偶极相互作用形成富阴离子溶剂化结构,其SEI/CEI展现出高稳定性,从而延长电池的使用寿命(图4j)。
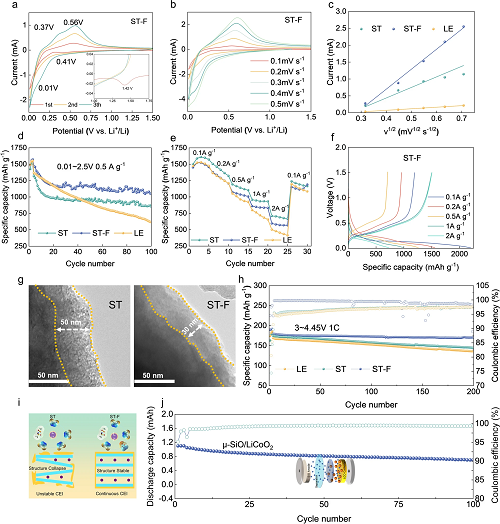
图4. Li|SiO,Li|LiCoO₂和SiO|LiCoO₂电池的电化学性能:(a)ST-F电解液的CV曲线;(b)在ST-F电解液中不同扫速下的SiO阳极CV曲线;(c)峰电流与扫描速度平方根之间的线性关系;(d)在0.5 A g⁻¹下Li|SiO电池的循环性能;(e)Li|SiO电池倍率性能测试;(f)在0.1 A g⁻¹至2 A g⁻¹下ST-F电解液中SiO阳极的充放电曲线;(g)使用丁二腈基电解液的循环后SiO阳极的TEM图像;(h)在1 C下Li|LiCoO₂电池的循环性能;(i)ST/ST-F电解液作用于LiCoO₂的示意图;(j)0.5 C下SiO|LiCoO₂电池的循环性能。
利用同步辐射X射线计算机断层扫描技术分析和量化循环的SiO中不同组分的分布。利用从多个区域获取的数据构建SiO阳极的3D微观结构(图5a)。在图5b中,ST-F电解液中循环的SiO阳极的空隙(3.9%)比ST电解液的空隙(10.6%)显著减小,这表明在ST-F电解液形成的SEI能够有效地减缓体积膨胀。在ST电解液中循环的SiO的F 1s光谱显示出弱的-CF₃信号(688.7 eV)(图5c),归因于TFSI⁻的分解。Li 1s光谱也存在LiₓN(54.7 eV)和LiF(55.1 eV)的信号,表明在ST-F电解液中SiO阳极上富含FEC/阴离子衍生的SEI的形成。C 1s光谱还显示了各种含碳组分的信号(图5e)。光学显微镜实验证明,与ST电解液相比,在ST-F电解液中循环后的SiO电极呈现出更平坦、更光滑的形貌(RZ=3.921),这归因于形成了更密集、更均匀的阴离子衍生SEI(图5f)。在ST电解液中,电解液在SiO阳极上持续发生还原分解,导致SEI主要由-CF₃、O-C=O、C-O、LiₓN和其他物质组成(图5g)。此外,在ST-F电解液中循环的SiO阳极表面还展现出更多的有机-无机成分,包括-CF₃、LiF和LiₓN,这些成分能够有效地抑制持续的体积膨胀和破裂。
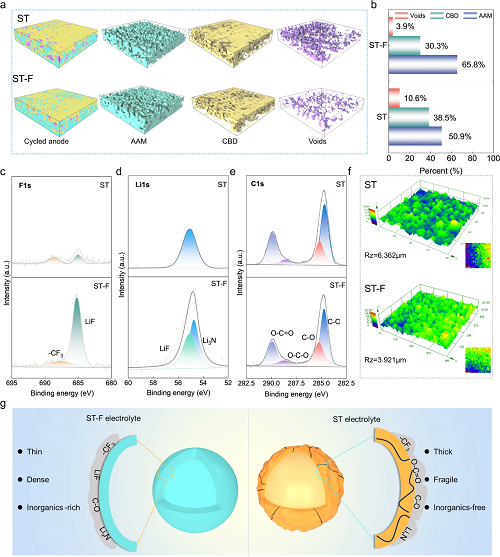
图5. 循环SiO阳极组分分布和表面化学性质分析:(a)同步辐射X射线计算机断层扫描测试;(b)各组分的百分比;循环后SiO阳极的XPS光谱分析:(c) Li 1s;(d)F 1s;(e)C 1s;(f)经ST/ST-F电解液循环后SiO阳极的粗糙度测试;(g)丁二腈基电解液中SiO阳极表面SEI成分的示意图。
III 总结总的来说,该工作报道了一种新型的丁二腈基电解液,旨在增强4.45 V SiO|LiCoO₂电池的长期循环性能,其具有阴离子主导的溶剂化结构。通过DFT计算和MD模拟,以调节丁二腈基电解液中的微观相互作用力(离子-离子、离子-偶极和偶极-偶极相互作用),使得即使使用较低浓度的锂盐,也能构建富含阴离子的溶剂化结构。通过引入弱溶剂化溶剂FEC,可以在分子水平上精确调控丁二腈基电解液中的Li⁺溶剂化结构,实现比LE电解液更高的离子电导率和迁移数。这解决了LE电解液中普遍存在的溶剂主导的溶剂化结构所带来的问题。Micro-CT展示了富含阴离子衍生的SEI能够有效抑制SiO在合金化/去合金化过程中不可逆的体积膨胀。这些发现充分证明了丁二腈基电解液促进高比容量硅基锂离子电池的商业化应用潜力。
作者简介

娄帅锋
本文通讯作者
哈尔滨工业大学 教授
▍主要研究领域
主要研究方向为铌基材料与快充电池、固态电池与智能分析、结构电池与力学等。
▍个人简介
哈尔滨工业大学教授,博士生导师。以第一/通讯作者发表SCI论文50余篇,包括Nat. Commun. (2)、JACS (2)、Chem、Adv. Mater.、Matter (2)、Energy Enviorn. Sci.、Nano Lett.、Adv. Funct. Mater. (2)等期刊,出版英文专著一部,主持国家自然科学基金(面上/青年)、国家重点研发计划子课题、黑龙江省优秀青年基金等项目,获黑龙江省自然科学奖、中国新锐科技人物知社特别奖,入选哈工大青年拔尖人才选聘计划、中国科协青年人才托举工程等。
▍Email:shuaifeng.lou@hit.edu.cn
撰稿:原文作者
编辑:《纳微快报(英文)》编辑部
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2023 JCR IF=31.6,学科排名Q1区前3%,中国科学院期刊分区1区期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 哈工大娄帅锋等:调控离子-偶极作用打破溶剂主导效应,突破一氧化硅阳极的长循环寿命
 河南大学武四新等:氢的光电特性调控和缺陷钝化作用实现高效银取代铜锌锡硫硒光伏器件
河南大学武四新等:氢的光电特性调控和缺陷钝化作用实现高效银取代铜锌锡硫硒光伏器件 NML文章集锦| 钙钛矿太阳能电池(四)
NML文章集锦| 钙钛矿太阳能电池(四) NML文章集锦| 钙钛矿太阳能电池(一)
NML文章集锦| 钙钛矿太阳能电池(一) 全南国立大学Chan-Jin Park等:在自支撑多孔石榴石骨架下原位聚合以构建高性能复合固体电解质
全南国立大学Chan-Jin Park等:在自支撑多孔石榴石骨架下原位聚合以构建高性能复合固体电解质