研究背景
电催化二氧化碳还原(CO₂RR)过程中各种中间体的吸附能具有很强的线性关系,限制了活性的进一步提升。本文构造了一个原子分散的Mo-Fe双原子对锚定在氮掺杂碳载体上。Mo-Fe双原子位点对*COOH中间体的“桥式”吸附方式增加了*COOH的吸附能。同时Mo和Fe原子之间的轨道耦合导致金属位点的电子离域,有利于*CO中间体的脱附。该策略打破了中间体吸附能之间的强相关性,使得该催化剂表现优于大多数同类催化剂的CO₂RR本征活性(TOF为3336 h⁻1),优异的CO选择性(FECO=95.96%)和良好的稳定性。
Atomic Dispersed Hetero-Pairs for Enhanced Electrocatalytic CO₂ Reduction
Zhaoyong Jin, Meiqi Yang, Yilong Dong, Xingcheng Ma, Ying Wang, Jiandong Wu, Jinchang Fan, Dewen Wang, Rongshen Xi, Xiao Zhao, Tianyi Xu, Jingxiang Zhao*, Lei Zhang*, David J. Singh, Weitao Zheng*, and Xiaoqiang Cui*
Nano-Micro Letters (2024)16: 4
https://doi.org/10.1007/s40820-023-01214-2
本文亮点
1. 成功设计并构建了一种独特的原子分散的Mo-Fe双原子对锚定在氮掺杂碳载体上。
2. 该策略通过同时调节*COOH吸附能和*CO解吸能,打破了电催化CO₂还原的线性相关束缚。
3. 制备的MoFe-N-C催化剂具有高本征活性(TOF为3336 h⁻1),高CO选择性(CO法拉第效率在-0.6 V下能达到95.96%)和良好的稳定性。
内容简介
电化学CO₂RR涉及多种中间体,不同中间体的吸附能的高度相关性阻碍了催化活性的优化。打破这种限制有望显著改善CO₂RR的本征活性,但仍然面临巨大的挑战。吉林大学崔小强教授、郑伟涛教授课题组与哈尔滨师范大学赵景祥教授课题组合作,首次通过构建一个独特的由原子分散的Mo-Fe双原子位点来解决这个难题。结果表明,Mo-Fe双原子位点通过对*COOH中间体的“桥式”吸附提高了*COOH中间体的吸附能。同时,Mo-Fe双原子之间的d-d轨道耦合导致电子离域,有利于*CO中间体的脱附。该策略突破了中间体吸附能之间线性关系的限制,该体系表现出由于优于绝大多数同类催化剂的本征活性、选择性和稳定性。
图文导读
I 原子分散的Mo-Fe异质双原子催化剂的制备及形貌分析
原子分散的Mo-Fe异质双原子催化剂(MoFe-N-C)的制备如图1A所示。通过改进的两步退火策略制备了锚定在氮掺杂碳载体上的原子分散的Mo-Fe双原子对。图1B的EPR光谱证明金属原子的成功锚定使EPR空位信号减弱。结合图1C的高角度环形暗场扫描透射电子显微镜图片(HAADF-STEM)和图1F-J的EDS Mapping证据表明观察到的大量成对出现的双金属位点为Mo-Fe原子对。图1E-F显示Mo-Fe原子对中Mo与Fe之间的统计距离为2.23±0.32 Å。
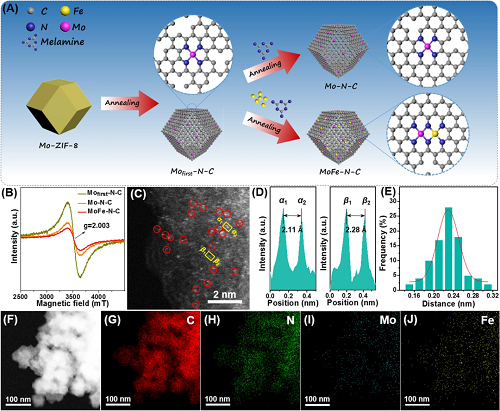
图1. (A)MoFe-N-C催化剂的制备示意图;(B)EPR光谱;(C)高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图片;(D)图(C)中黄色方框内得到的亮点的亮度分布图;(E)观测到的Fe-Mo双原子对的统计距离。(F)-(J)C、N、Mo和Fe元素在MoFe-N-C上的分布。
II 催化剂精细结构表征
Fe和Mo的X射线近边吸收结构(图2A-B)表明MoFe-N-C中Mo-Fe原子对的形成使Mo和Fe的价态同时升高。如图2C-D所示,在MoFe-N-C傅里叶变换后的X射线吸收光谱中同时观察到Mo-N键、Fe-N键和Mo-Fe键的存在,证明了Mo-Fe原子对以金属-氮的配位形式锚定在碳载体上。这一结果同样在小波变换的结果中得到证明(图2E)。此外MoFe-M-C催化剂中的Fe-Mo键与金属的Fe-Fe和Mo-Mo键之间都具有较大差异,证明MoFe-M-C中不存在金属原子的团聚。如图2F所示,扩展X射线吸收光谱的拟合结果与图2G的模型展示的结构吻合,即Mo-Fe原子对和周围6个N原子配位形成一个MoFe-N₆位点。
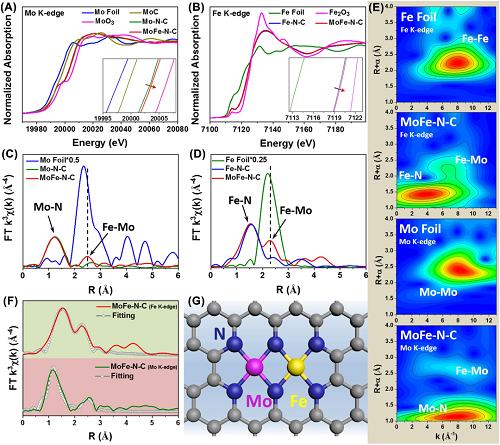
图2. (A)Mo K-edge和(B)Fe K-edge的X射线近边吸收光谱;傅里叶变换后的(C)Mo K-edge和(D)Fe K-edge的X射线吸收结构;(E)小波变换;(F)MoFe-N-C的扩展X射线吸收光谱的拟合曲线;(G)MoFe-N-C的结构模型。
III 催化剂性能的表征
测试了催化剂电化学CO₂RR的性能。线性扫描伏安曲线(图3A)表明MoFe-N-C具有最大的电化学活性。图3B-D对产物的法拉第效率、部分电流密度和周转率进行评估,发现MoFe-N-C展现了最佳的CO选择性和最大的CO₂RR反应活性。CO₂气体化学吸脱附曲线证明Mo单原子催化剂(Mo-N-C)和MoFe-N-C对CO₂有较强的化学吸附,有利于CO₂的活化,而Fe单原子催化剂(Fe-N-C)的CO₂吸附能力较弱。CO气体化学吸脱附曲线证明Mo-N-C较强的CO吸附可能是导致其选择性较低的原因,而MoFe-N-C的优异性能可能来源于其同时具有较强的CO₂吸附能力和较弱的CO吸附能力。
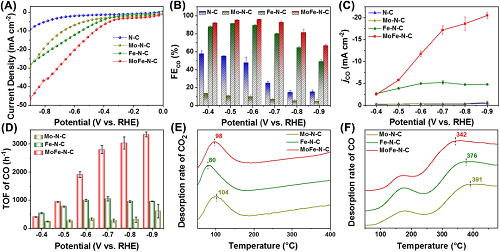
图3. (A)线性扫描伏安曲线;(B)产物CO的法拉第效率;(C)CO的部分电流密度;(D)周转率;(E)CO₂气体和(F)CO气体的程序升温脱附曲线。
IV 催化剂机理的探讨
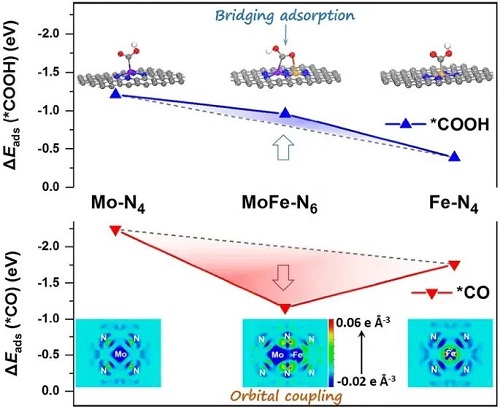
如图4A所示,构建了CO₂RR反应过程中催化位点的结构模型,发现MoFe-N-C中的Mo和Fe原子位点分别与*COOH中间体的C和O原子相连。这种特有的“桥式”吸附结构有利于电子转移,增加了*COOH中间体的吸附能。图4B显示MoFe-N-C的MoF-N₆位点虽然具有较大的*COOH吸附能,但是*CO中间体的吸附能较小。态密度分析(图4C)显示MoFe-N-C中Mo 4d轨道和Fe 3d轨道在费米能级附近发生强烈的共振,表明Mo-Fe之间的d-d轨道耦合。这种轨道耦合导致了金属位点的电子离域,即金属位点的电子向配位的氮原子转移,这有利于*CO中间体的脱附。图4D中的吉布斯自由能表明相比于其他催化剂MoFe-N-C的CO₂RR的能垒最低。CO₂RR和HER热力学极限电势差(图4E)则表明MoFe-N-C具有更好的CO₂RR选择性。以上结果表明MoFe-N-C催化性能提升的主要原因是原子分散的Mo-Fe双原子对打破了不同中间体吸附能的线性关系并降低了CO₂RR能垒。
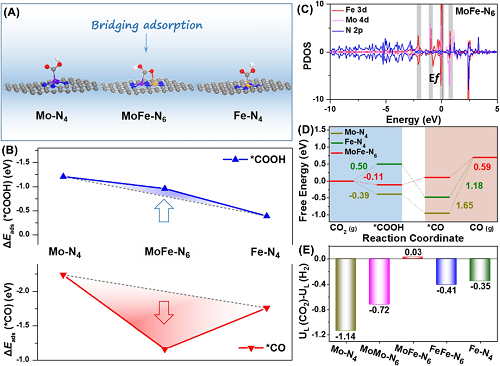
图4. (A)吸附*COOH中间体的催化剂结构模型;(B)*COOH和*CO中间体的吸附能对比;(C)MoFe-N-C中Mo 4d、Fe 3d和N 2p轨道的部分态密度;(D)吉布斯自由能;(E)CO₂RR和HER热力学极限电位差。
作者简介

本文通讯作者

本文通讯作者
低维纳米材料电催化性能的理论研究。
▍Email:zhaojingxiang@hrbnu.edu.cn

本文通讯作者
超硬薄膜材料、功能薄膜材料、石墨烯等碳纳米材料、储能电极材料及材料计算与模拟。
▍Email:wtzheng@jlu.edu.cn
关于我们

Nano-Micro Letters《纳微快报(英文)》是上海交通大学主办、在Springer Nature开放获取(open-access)出版的学术期刊,主要报道纳米/微米尺度相关的高水平文章(research article, review, communication, perspective, highlight, etc),包括微纳米材料与结构的合成表征与性能及其在能源、催化、环境、传感、电磁波吸收与屏蔽、生物医学等领域的应用研究。已被SCI、EI、PubMed、SCOPUS等数据库收录,2022JCR影响因子为 26.6,学科排名Q1区前5%,中科院期刊分区1区TOP期刊。多次荣获“中国最具国际影响力学术期刊”、“中国高校杰出科技期刊”、“上海市精品科技期刊”等荣誉,2021年荣获“中国出版政府奖期刊奖提名奖”。欢迎关注和投稿。
Web: https://springer.com/40820
E-mail: editor@nmlett.org
Tel: 021-34207624
如果文章对您有帮助,可以与别人分享!:Nano-Micro Letters » 吉林大学崔小强等:原子分散的异质双原子催化剂增强电催化CO₂RR
 西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化
西交戴正飞和西工大瞿永泉等:镍功能化的黑磷烯复合材料,实现高效碱性析氢/析氧电催化